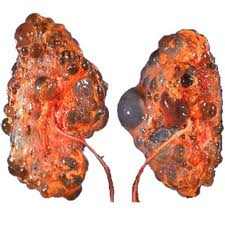
Polycystic kidney disease is a kidney disorder passed down through families in which many cysts form in the kidneys, causing them to become enlarged.
Causes, incidence, and risk factors
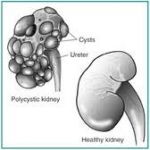 Polycystic kidney disease (PKD) is passed down through families (inherited), usually as an autosomal dominant trait. If one parent carries the gene, the children have a 50% chance of developing the disorder.
Polycystic kidney disease (PKD) is passed down through families (inherited), usually as an autosomal dominant trait. If one parent carries the gene, the children have a 50% chance of developing the disorder.
Autosomal dominant PKD occurs in both children and adults, but it is much more common in adults. Symptoms often do not appear until middle age. It affects nearly 1 in 1,000 Americans. The actual number may be more, because some people do not have symptoms.
An autosomal recessive form of PKD also exists. It appears in infancy or childhood. This form is much less common than autosomal dominant PKS, but it tends to be very serious and gets worse quickly. It can cause serious lung and liver disease, end-stage kidney disease, and it usually causes death in infancy or childhood.
Persons with PKD have many clusters of cysts in the kidneys. What exactly triggers the cysts to form is unknown.
PKD is associated with the following conditions:
- Aortic aneurysms
- Brain aneurysms
- Cysts in the liver, pancreas, and testes
- Diverticula of the colon
As many as half of people with PKD have cysts on the liver. A family history of PKD increases your risk for the condition.
Pathophysiology
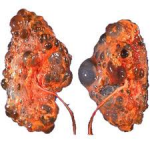
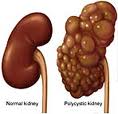
Appearance of kidney in polycystic kidney disease
Recent studies in fundamental cell biology of cilia and flagella using experimental model organisms such as the round worm Caenorhabditis elegans and the mouse Mus musculus have shed light on how PKD develops in human patients. Biochemist James Calvet writes: “These discoveries are a testament to the power of genetics and the importance of animal models. Who would have thought to look at cilia at all as a basis for ADPKD if the direction had not been pointed out by these genetic studies?”
All cilia and flagella are constructed and maintained, by the process of intraflagellar transport, a cellular function that is also essential for the insertion of proteins at specific sites along cilia and flagella membranes. These inserted membrane proteins can initiate environmental sensing and intracellular signaling pathways. They play a special role in the cilia of renal epithelial cells, and are thought to be critical for normal renal cell development and function and are sorted out and localized to the cilia of renal epithelial cells by the aforementioned intraflagellar transport mechanism. Ciliated epithelial cells line the lumen of the urinary collecting ducts and sense the flow of urine. Failure in flow-sensing signaling results in programmed cell death (which is also known as apoptosis) of these renal epithelial cells, producing the characteristic multiple cysts of PKD. PKD may result from mutations of signaling and environmental sensing proteins, or failure in intraflagellar transport.
Two PKD genes, PKD1 and PKD2, encode membrane proteins that localize to a non-motile cilium on the renal tube cell. Polycystin-2 encoded by PKD2 gene is a calcium channel that allows extracellular calcium ions to enter the cell. Polycystin-1, encoded by PKD1 gene, is thought to be associated with polycystin-2 protein and regulates polycystin-2’s channel activity. The calcium ions are important cellular messengers, which trigger complicated biochemical pathways that lead to quiescence and differentiation. Malfunctions of polycystin-1 or polycystin-2 proteins, defects in the assembly of the cilium on the renal tube cell, failures in targeting these two proteins to the cilium, and deregulations of calcium signaling all likely cause the occurrence of PKD.
Genetics
 As stated above, defects in two genes are thought to be responsible for ADPKD. In 85% of patients, ADPKD is caused by mutations in the gene PKD1 on chromosome 16 (TRPP1); in 15% of patients mutations in PKD2 (TRPP2) are causative.
As stated above, defects in two genes are thought to be responsible for ADPKD. In 85% of patients, ADPKD is caused by mutations in the gene PKD1 on chromosome 16 (TRPP1); in 15% of patients mutations in PKD2 (TRPP2) are causative.
PKD and the “two hit” hypothesis
The two hit hypothesis (aka Knudson hypothesis ) is often used to explain the manifestation of polycystic kidney disease later in life even though the mutation is present at birth. This term is borrowed from cancer research stating that both copies of the gene present in the genome have to be “silenced” before cancer manifests itself (in Knudson’s case the silenced gene was Rb1). In ADPKD the original “hit” is congenital (in either the PKD1 or PKD2 genes) and the subsequent “hit” occurs later in life as the cells grow and divide. The two hit hypothesis as it relates to PKD was originally proposed by Reeders in 1992. Support for this hypothesis comes from the fact that ARPKD patients develop disease at birth, and somatic mutations in the “normal” copy of PKD1 or PKD2 have been found in cyst-lining epithelia.